Biologic Scholar Winston Ewert Coauthors New Edition of The Design Inference
The second edition of The Design Inference was recently released. Winston Ewert from the Biologic Institute coauthored the book with William Dembski who wrote the first edition. The original book presented a general methodology for detecting design, which was the most sophisticated framework to date. The second edition represents the culmination of two decades of responding to critics and refining his model. Princeton University mathematician Sergiu Klainerman welcomed the book as follows:
Well argued and eminently readable… I don’t see how any open-minded scientist can ignore this important book.
The first edition of The Design Inference explained how design detection involves identifying patterns that are both highly improbable and specified. A common question raised by readers was the precise definitions for the terms specified and specification. In other words, what exactly makes a pattern specified or special? In many contexts, the answer is simple. For instance, being dealt four poker hands that were all royal flushes would be a very rare and special pattern pointing to someone cheating. Design detection in this example is easy because the category royal flush was already defined (aka prespecified) by the rules of the game. The situation is more challenging when categories of outcomes are not so clearly defined.
In the new edition, Dembski and Ewert greatly refined the concept of specifications. They defined a specification as a description that requires few words. The description must also only apply to a small proportion of possible outcomes, so the probability of any outcome falling within the specification must be sufficiently small. Patterns that are both identifiable by a short description and highly improbable are almost always designed.
Returning to the poker example, the four hands I mentioned could be described or specified by the three words four royal flushes, and the probability of four random hands falling within this specification is less than 1 chance in 100 billion trillion. The short description and very low probability indicate design.
In contrast, four typical hands of five cards would require a very long description, such as the following:
two of diamonds, pair of threes, pair of jacks; three fours, nine of hearts, ten of clubs; four of diamonds, pair of eights, nine of clubs, queen of clubs; three kings, pair of aces
The probability of four hands falling within this description is low, but the description length is very long. Many sets of four poker hands would fall within a description of that length. The low probability is offset by the large number of sets of four hands with descriptions that are that long. The probability of being dealt four hands with such a long description is not sufficiently low to conclude design.
Conversely, one could label four generic hands with the description random cards. The description is now short, but the probability is very high of four hands falling within this description. The short description is offset by the high probability. Similarly, the famous face on Mars can be described by the two words fuzzy face. Here again, the description length is short, but the probability of a photograph of the surface of a celestial body resembling a face at least as well as the Mars photo is sufficiently high to occur by chance.
ALT
The face on Mars (NASA/JPL)
The underlying logic of Dembski and Ewert’s methodology is profound. Identifying design requires a pattern being assigned by a mind a special significance. A short description meets this criterion since societies assign words or short phrases to that which they designate as special.
To illustrate, in the movie The Empire Strikes Back, Darth Vader declares to Luke Skywalker one of the most iconic lines in movie history: “I am your father.”
Darth Vader could specify himself by the single word father. The relationship between a father and son is designated by all societies as a very special relationship. Relatively few other people could be specified by such a short description. Consequently, the audience knew Luke’s meeting Vader was not an accident but by design.
In stark contrast, Dark Helmet in the movie Spaceballs tells Lonestar a far longer statement: “I am your father’s brother’s nephew’s cousin’s former roommate.”
The fact that Dark Helmet had to employ such a lengthy description to specify himself reflects that their relationship carried no significant meaning. Such a generic relationship could have occurred by chance. Dembski and Ewert detail how their methodology could be applied in many other contexts with equally reliable results.
Does Science Rule Out a First Human Pair? Geneticist Richard Buggs Says No
Dennis Venema, associate professor of biology at Trinity Western University, and Scot McKnight, professor of New Testament at Northern Seminary, have written a book, Adam and the Genome: Reading Scripture after Genetic Science, that addresses the question of human origins. Venema defends the standard scientific narrative on neo-Darwinism and human origins, while McKnight attempts to reconcile Christian theology with the science. Because the book argues that science disproves the existence of a historical Adam and Eve, it is worth responding to in some detail.
One of the claims Venema makes in his book is that the effective population size of our last common ancestor with chimps has never been fewer than 10,000. In fact, he equates the certainty of that statement with our certainty about heliocentrism. That is an extreme claim that needs justification. Assuming no bottlenecks, this would exclude the possibility of an original human pair as the progenitors of the human race.
Effective population size is very hard to determine. In fact, in some situations the effective population size cannot be determined (On the Meaning and Existence of an Effective Population Size P. Sjödin, I. Kaj, S. Krone, M. Lascoux and M. Nordborg (2005) Genetics 169: 1061–1070). So any estimates concerning effective population size should be taken a bit skeptically.
You don’t have to take my word for it. Richard Buggs, a British biologist who has published over thirty articles on genetics in journals such as Nature, Current Biology, Evolution, and Philosophical Transactions of the Royal Society, wrote to Venema to lay out his concerns in May of this year. Venema failed to respond to Buggs’s email so Buggs posted it online last week, and tweeted a link to it here. In his letter he comments on effective population size estimates:
As I am sure you know, effective population size is a measure of a population’s susceptibility to drift, rather than an attempt to measure census population size. I would be very hesitant to rely too heavily on any estimate of past effective population size.
Could we have begun with a population of 10,000 and then had a bottleneck of two? Obviously this estimate is relevant for human origins, since a bottleneck of two would indicate some sort of unique beginning. Venema says a bottleneck of two is impossible based on the levels of human genetic diversity present today.
If a species were formed through such an event [by a single ancestral breeding pair] or if a species were reduced in numbers to a single breeding pair at some point in its history, it would leave a telltale mark on its genome that would persist for hundreds of thousands of years — a severe reduction in genetic variability for the species as a whole.
Buggs responds:
It is easy to have misleading intuitions about the population genetic effects of a short, sudden bottleneck. For example, Ernst Mayr suggested that many species had passed through extreme bottlenecks in founder events. He argued that extreme loss of diversity in such events would promote evolutionary change. His intuition about loss of diversity in bottlenecks was wrong, though, and his argument lost much of its force when population geneticists (M. Nei, T. Maruyama and R. Chakraborty 1975 Evolution, 29(1):1-10) showed that even a bottleneck of a single pair would not lead to massive decreases in genetic diversity, if followed by rapid population growth. When two individuals are taken at random from an existing large population, they will on average carry 75% of its heterozygosity (M. Slatkin and L. Excoffier 2012 Genetics 191:171–181). From a bottleneck of a single fertilised female, if population size doubles every generation, after many generations the population will have over half of the heterozygosity of the population before the bottleneck (Barton and Charlesworth 1984, Ann. Rev. Ecol. Syst. 15:133-64). If population growth is faster than this, the proportion of heterozygosity maintained will be higher.
This means that a single pair of individuals can carry a great deal of heterozygosity with them through a bottleneck, provided they come from an ancestral population with high diversity and undergo rapid population growth after the bottleneck. They will pass most of that diversity on to the population they found, so long as population grows rapidly.
Buggs goes on to discuss the relative genetic diversity of chimpanzees at 5.7 million SNPs (single nucleotide polymorphisms) and humans at 3.1 million (Prado-Martinez et al 2013 Nature). He makes the point that if a pair of chimpanzees were moved to an isolated region where rapid population growth was possible, the new population would have similar levels of genetic variability to modern humans.
He explains:
I am not stating these figures because existing populations of chimpanzee gave rise to modern humans, but simply to show that it is hard to see how overall levels of SNP diversity and heterozygosity in modern humans could exclude the possibility of a past bottleneck of two individuals.
On top of this, we need to add in the fact that explosive population growth in humans has allowed many new mutations to rapidly accumulate in human populations, accounting for many SNPs with low minor allele frequencies (A. Keinan and A. G. Clark (2012) Science 336 (6082): 740-743).
PSMC (pairwise sequentially Markovian coalescent) analysis is a method used by population geneticists to try to recover the history of human populations. Venema discusses it in his book. Unfortunately he cannot use it to prove that a sudden, short bottleneck never happened. In the original Li and Durbin 2011 paper describing PSMC analysis, the authors note that an instantaneous reduction in population size was not detected as such, but was instead “spread over several preceding tens of thousands of years in the PSMC reconstruction.” Continued work in Beth Shapiro’s lab also indicates that the PSMC method cannot accurately reconstruct sharp bottlenecks.
Buggs sums up his problems with Venema’s analysis:
In general, I am concerned that the studies you cite did not set out to test the hypothesis that humans have passed through a single-couple bottleneck. They are simply trying to reconstruct the most probable past effective population sizes of humans given the standard assumptions of population genetic models. I personally would feel ill at ease claiming that they prove that a short sudden bottleneck is impossible. [Emphasis added.]
Other scientistshaveargued against Venema’s position by disputing key assumptions, including the idea of common descent. Buggs’s argument is valid, though, whether or not common descent is true. In fact, he has told me he is assuming common ancestry, but arguing that we can still have come from a single couple as ancestors.
What is needed is a model that does not rely on the standard assumptions of population genetics. Recently Hössjer, Gauger, and Reeves have proposed such an alternative model. The model, when programmed, will be available for use by anyone to test the effects of various starting conditions, like population size, the degree of gene flow, recombination and mutation rates, mate choice effects, and ultimately the effects of selection. The outcomes of various scenarios can be tested and compared to summary statistics from modern populations, in order to determine which scenarios best explain current populations.
Finally, given Buggs’s critique, Venema’s claims that our starting population was at least 10,000, and that we could not have come from just two, seem unjustified. Certainly Venema cannot claim these are facts as sure as heliocentrism. He should at least acknowledge the facts above, and soften his claims. Further revision may be necessary following the results from Hössjer, Gauger, and Reeves’s model. We shall see.
There is a whole school of research aimed at showing we are not so separate from chimps, and even that they are already on the way up. In a recent BBC article by Colin Barras the focus was on the use of stone tools, and the bold claim that chimps and some monkeys are already in the Stone Age because of their use of rocks as tools.
Some of the reports of stone use appear valid. Capuchins and chimpanzees are all known to use stones to crack open food, and the technique appears to go back thousands of years. But then, no one is disputing that some animals use simple tools. Even otters use stones to break open clam shells.
Then there is the social side. Another BBC article, this one by Melissa Hogenboom, describes ways that chimps show empathy and social awareness, the ability to read facial expressions, to hide things from others, and to share. But then those things are not so unique–anyone who has lived with dogs knows how acutely sensitive to social cues and facial expressions they are, and how they seem to show guilt over bad behavior. They can even share–or be greedy, depending on the dog’s temperament.
And of course, she describes how chimps have the capacity for language. Translation: chimps that have lived and trained with humans can have a vocabulary of 500 words, and can understand thousands more.
But is this a sign they are on the road to becoming like us?
In a further BBC article, Hogenboom lays out the argument against the idea that there is little difference between us and chimps. She quotes Ian Tatersall, a paleoanthropologist at the American Museum of Natural History in New York: “Obviously we have similarities. We have similarities with everything else in nature; it would be astonishing if we didn’t. But we’ve got to look at the differences,”
Hogenboom writes:
When you pull together our unparalleled language skills, our ability to infer others’ mental states and our instinct for cooperation, you have something unprecedented. Us.
‘Just look around you,’ Tomasello [of the Max Planck institute for Evolutionary Anthropology in Leipzig, Germany,] says, ‘we’re chatting and doing an interview, they (chimps) are not.’
We have our advanced language skills to thank for that. We may see evidence of basic linguistic abilities in chimpanzees, but we are the only ones writing things down.
We tell stories, we dream, we imagine things about ourselves and others and we spend a great deal of time thinking about the future and analyzing the past.
Not to mention the Brandenburg Concertos. Calculus. Hamlet. The Pythagorean theorem. The discovery of the periodic table. Einstein’s theory of relativity. All works of genius.
In her first article I think Hogenboom overplays the similarities between chimps and humans, just as Barras exaggerates their tool-making ability. Both authors accept the standard Darwinian view, however. Perhaps that accounts for their need to have a story of underlying similarity.
Ms. Hogenboom attributes all this to our big brains in her second article. She does admit that nobody knows how our brains came to be big. Neither does anyone have any solid explanation for how the anatomical, developmental, and physiological differences between us and chimps came to be.
At least Ms. Hogenboom acknowledges there might be a difficulty in explaining things.
Nylonase: Move On, Nothing to See Here, says Theistic Evolutionist
Ann Gauger
The supposed sudden emergence of the enzyme nylonase has been a chief talking point for the power of evolution for many years, and it has made its appearance multiple times in Dennis Venema’s series of posts, “Letters to the Duchess,” at the theistic evolutionist website BioLogos. Here’s how Venema, Fellow of Biology, described it in a post last year:
[Nylonase] arose from scratch as an insertion mutation into the coding sequence of another gene. This insertion simultaneously formed a “stop” codon early in the original gene (a codon that tells the ribosome to stop adding amino acids to a protein) and formed a brand new “start” codon in a different reading frame. The new reading frame ran for 392 amino acids before the first “stop” codon, producing a large, novel protein. As in our example above, this new protein was based on different codons due to the frameshift. It was truly “de novo” — a new sequence.
Why was this supposed to be important? Venema continued:
In order for proteins to function, they need to fold up into stable shapes — “protein folds”. Meyer claims, based on the work of his colleague Douglas Axe, that stable protein folds are vanishingly rare — on the order of only one in 10 to the 77th power. As such, Meyer argues, evolution cannot find the needle (a functional protein) in the haystack (the vast number of functionless possibilities) … All of this hinges, of course, on just how accurate Axe’s estimate is: are functional proteins really that rare?…
So if nylonase arose by frameshift mutation, via a single base insertion, producing a novel sequence and a stable, functional fold, it would appear that functional, stable folded proteins are not rare. Venema said:
…this brand-new protein [nylonase] folded into a stable shape, and acted as a weak nylonase. Later duplications, mutations and selection would make a very efficient nylonase from this starting point. Additionally, the three-dimensional structure of the protein has been solved using X-ray crystallography, a method that gives us the precise shape of the protein at high resolution. Nylonase is chock full of protein folds — exactly the sort of folds Meyer claims must be the result of design because evolution could not have produced them even with all the time since the origin of life.
Put another way, if only one in 10 to the 77th proteins are functional, there should be no way that this sort of thing could happen in billions and billions of years, let alone 40. Either this was a stupendous fluke (and stupendous isn’t nearly strong enough of a word), or evolution is in fact capable of generating the information required to form new protein folds.
And if this can happen in 40 years, what might millions of years of evolution produce? [Emphasis added.]
Venema was merely repeating the old story that has long circulated around many anti-ID websites. It goes back to 1984, when Susumu Ohno first proposed a frameshift mutation as the cause of the new nylonase enzyme.
…what would truly settle the issue is a concrete example of a “truly new” gene coming into being — one that is not a duplicate of a preexisting protein sequence — that nonetheless has a new function and new protein folds. If scientists could observe such an event, then it would indicate that Axe’s math (and Meyer’s use of it) is not a reliable estimate for the prevalence of functional protein folds.
Now, here’s where I come in. It seemed highly unlikely to me that a stable, functional protein fold could arise by frameshift mutation, unless the sequence was designed that way. Among geneticists frameshift mutations are called “nonsense” mutations because they typically result in scrambled non-functional code. In fact, this view of frameshift mutations was pretty universal until we began to encounter what looked like frameshifts in sequenced genomes.
So I began to dig in the literature. What I found was a major block across the road of the old nylonase story. To get the full picture, I’d suggest reading this post I wrote, “The Nylonase Story: When Imagination and Facts Collide,” back in May. It is based on the paper by Negoro et al. describing nylonase’s structure. The paper makes clear that nylonase was a variant of a pre-existing β-lactamase fold, not a novel protein fold. In fact, among the different kinds of β-lactamases, nylonase most resembled carboxylesterases. It was then found that nylonase had previously undetected carboxylesterase activity.
In other words, nylonase had started out as a carboxylesterase, then evolved the ability to degrade nylon byproducts. The title of the paper, and its Abstract, should make the truth obvious:
X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer-degrading enzyme.
Here, we propose that amino acid replacements in the catalytic cleft of a preexisting esterase with the β-lactamase fold resulted in the evolution of the nylon oligomer hydrolase [nylonase].
Nylonase did not have a frameshifted new protein fold. It was a pre-existing fold with activity characteristic of its fold type. No novel protein fold had emerged. And no frameshift mutation was required to produce nylonase. To quote my post:
Where did the nylon-eating ability come from? Carboxylesterases are enzymes with broad substrate specificities; they can carry out a variety of reactions. Their binding pocket is large and can accommodate a lot of different substrates. They are “promiscuous” enzymes, in other words. Furthermore, the carboxylesterase reaction hydrolyzes a chemical bond similar to the one hydrolyzed by nylonase.
“Our studies demonstrated that among the 47 amino acids altered between the EII and EII’ proteins, a single amino acid substitution at position 181 was essential for the activity of 6-aminohexanoate-dimer hydrolase [nylonase] and substitution at position 266 enhanced the effect.”
So. This is not the story of a highly improbable frame-shift producing a new functional enzyme. This is the story of a pre-existing enzyme with a low level of promiscuous nylonase activity, which improved its activity toward nylon by first one, then another selectable mutation. In other words this is a completely plausible case of gene duplication, mutation, and selection operating on a pre-existing enzyme to improve a pre-existing low-level activity, exactly the kind of event that Meyer and Axe specifically acknowledge as a possibility, given the time and probabilistic resources available. Indeed, the origin of nylonase actually provides a nice example of the optimization of a pre-existing fold’s function, not the innovation or creation of a novel fold.
The data described in the Negoro et al. and Kato et al. papers build a brick wall right across the path of Venema’s argument. And sadly, that roadblock pre-existed Venema’s posts. If he had looked carefully he would have seen it.
What do you suppose Venema’s reaction was to this collision of imagination and fact? He responded by saying, essentially, “Move on, nothing to see here.” A combination of selective retelling, sleight of hand, redirection, and downgrading the importance of the nylonase story were his main techniques. Let’s take Venema’s final post on nylonase a bit at a time to see how he did it. Dated May 18, 2017, it bears the headline:
He begins with the canonical story, simply restated. No mention of the contrary facts.
In previous posts in this series, we’ve explored the claim made by the Intelligent Design (ID) movement that evolutionary mechanisms are not capable of generating the information-rich sequences in genes. One example that we have explored is nylonase — an enzyme that allows the bacteria that have it to digest the human-made chemical nylon, and use it as a food source. As we have seen, nylonase is a good example of a de novo gene — a gene that arose suddenly and came under natural selection because of its new and advantageous function. Since nylonase is a folded protein with a demonstrable function, it should be beyond the ability of evolution to produce, according to ID.
But Venema did notice I was writing about the subject. I assume he read what I wrote.
The implications of nylonase for ID arguments are clear, and they have caught the notice of several ID supporters. In recent weeks a number of posts on the subject have appeared on ID websites. ID biologist Anne [sic] Gauger, for example, is writing a series of posts in an attempt to rebut the evidence that nylonase is in fact a de novo gene. Her motivation for this work is clear. [Emphasis added.]
But at this point, oddly enough, rather than describe and refute my arguments, he changes the subject. He spends the rest of the post discussing a new paper that deals with other data regarding the ease with which random sequences benefit bacteria. He only mentions nylonase to dismiss it, in the closing paragraphs.
As we can see, the issue at hand is not so much the specific origins of nylonase, but rather the relative ease by which new, functional proteins can come into existence. If it is easy to evolve them, ID advocates are wrong. If new protein functions are vanishingly rare and inaccessible to evolution, ID would be strongly supported. With nylonase, we are dealing with events that happened in the past, so our inferences are limited to working with the evidence we have in the present.
So, we can see that the nylonase issue is something of a distraction — a missing of the forest to focus on one particular tree. Even if this particular example could have an alternate explanation, as Gauger argues, the problems for ID do not go away. [Emphasis added.]
This is a classic tactic. When shown to be wrong about nylonase, Venema changes the subject. Now nylonase is something that happened in the past, he says, implying we can’t reliably make inferences about it. He even says that nylonase is a “distraction” — despite his own repeated use of the nylonase story in trying to argue against Stephen Meyer and Doug Axe.
So as to acknowledge what Venema did talk about, I offer a few comments about the issue of functional, random sequences. Venema described the results of a recent study where a verysurprising number of random sequences were reported to confer a fitness advantage on cells expressing them. I have my doubts about these results and will address them on another occasion. But I am not the only one who should be having doubts. Whatever those proteins or RNAs are doing, it is unlikely to be via a stable functional fold.
François Jacob wrote a famous essay, “Evolution and Tinkering,” in which he said, speaking of the time when the first primitive cells had emerged:
New functions developed as new proteins appeared. But these were merely variations on previous themes. A sequence of a thousand nucleotides codes for a medium-sized protein. The probability that a functional protein would appear de novo by random association of amino acids is practically zero. In organisms as complex and integrated as those that were already living a long time ago, creation of entirely new nucleotide sequences could not be of any importance in the production of new information. [Emphasis added.]
I am not denying the existence of de novo genes — ORFan genes, for example, are undeniable. What Doug Axe, Stephen Meyer, and I say is that the probability of the appearance of de novo functional protein folds by random association of amino acids is practically zero.
Editor’s note: Nylon is a modern synthetic product used in the manufacturing, most familiarly, of ladies’ stockings but also a range of other goods, from rope to parachutes to auto tires. Nylonase is a popular evolutionary icon, brandished by theistic evolutionist Dennis Venema among others. In a series of three posts, of which this is the third, Discovery Institute biologist Ann Gauger takes a closer look. Look here for the first and second posts.
Returning to the story of the nylonase gene and the problem of where new information comes from, I’d like to make the point that there is a reason that molecular geneticist and evolutionary biologist Susumu Ohno made his hypothesis about a frame-shift having produced nylonase. Ohno is famous for his hypothesis that gene duplication and recruitment are the chief means by which “new” proteins are made — he wrote a famous book about it.
But he also knew that copying and tinkering weren’t enough, that there had to be a way to generate genuine de novo information, brand new coding sequence for genuinely new proteins, in order to account for all the diversity of information that must have been necessary as life became more complex. New proteins had to come from somewhere.
Ohno had an idea. He thought coding sequences made up of oligomeric repeats might allow there to be several alternate ways to read the same sequence. For an explanation of alternate reading frames, see my earlier post, “The Nylonase Story: How Unusual Is That?”
As a potential example, Ohno proposed nylB, the gene for nylonase. This gene has certain characteristics that make it plausible that a frameshift could have occurred, characteristics I described in that second post in this series, such as nylB’s sequence being GC-rich and deficient in TAs. These two characteristics reduce the chances of having stop codons, in any frame.
Ohno thus proposed that nylonase arose after a frameshift mutation in a perhaps nonfunctional, prior-coding sequence, resulting in an entirely new coding sequence with nylonase activity. The only reason Ohno could make this proposal was because nylB, the gene that codes for nylonase, has at least two potential open reading frames in the forward direction — the hypothetical “original” one proposed by Ohno from before any hypothetical T insertion took place, and the actual one that codes for nylonase now.
Ohno published his paper in 1984. In 1992, Yomo et al. noticed that one frame in the antisense direction of nylonase has no stop codons either. It also lacks a start codon, though, so Yomo et al. called it a non-stop frame (NSF) instead of an open reading frame (ORF). The probability of finding a DNA sequence with an ORF on the sense strand and a full NSF on the antisense strand are small. But surprisingly, not only does nylB have an NSF on the antisense stand, nylBhas another fully overlapping NSF in the forward direction. That’s two NSFs plus the actual ORF for nylonase (I’m not counting the hypothetical frame-shifted “original” ORF, since that frame actually has several intervening stops. (See “The Nylonase Story: When Facts and Imagination Collide.”) That means nylB has no stop codons in three out of six frames.
The chances of avoiding a stop codon in three out of six frames are very low. Our simulation (described in the previous post) showed that the probability is very small indeed. For an ORF 900 nucleotides long to have two NSFs at 70 percent GC is 9 out of 28,603 or .0003. (See there for details.) If these figures are recast to include the total number of random trials required to get an ORF of the proper length and GC content in the first place, and then with two NSFs, then the probability would be nine out of ten million trials, or 0.0000009. No organisms have ten million genes (we only have about twenty thousand), and Flavobacterium certainly doesn’t. But it’s not outside the realm of possibility that such sequences should exist by pure chance somewhere. After all, nylB does. But take the following into consideration.
In addition, beyond the first appearance of such a sequence, there would also need to be some way to prevent random mutation from introducing any stop codons over evolutionary time, in any of the three open frames. Purifying selection would normally be invoked in such a case. Organisms that develop harmful mutations in genes that encode functional gene products — things that are important for the organism’s survival — are less successful at reproducing, and so organisms carrying harmful mutations tend to disappear from the population (they are sickly or dead). However, purifying selection by definition has no effect on non-functional sequences. The fact that stops are prevented from accumulating in nylB NSFs implies that all three frames are functional. No function has been reported for the NSFs, however. They have no ATGs in the vicinity and so may be non-coding (though it must be acknowledged there are alternate start codons in the vicinity). In addition, it has been reported that the pOAD2 plasmid on which nylB is located is non-essential. It can be cleared from its host with no effect, except the loss of the ability to degrade nylon.
One possibility is that nylB has a secondary DNA or RNA-based function that requires its sequence to be nearly completely conserved. It would have to be a very specific sequence requirement to prevent the accumulation of stop codons in three frames, though. We get a hint that the cause is not sequence specificity, because the nylB and nylB′ genes of Flavobacterium differ by 47 amino acids, and the nylBgene of Pseudomonas has only about 35 percent identity according to reports, yet all three lack stops in the anti-sense frames in addition to their coding sequence (based on available sequence information).
Yomo et al., who first reported the anti-sense NSF in nylB, were amazed and puzzled by the existence of anti-sense NSFs in nylB genes of multiple species.
The probability of the presence of these NSFs on the antisense strand of a gene is very small (0.0001-0.0018) [we observed .0001]. In addition, another gene for nylon oligomer degradation [Pseudomonas nylB] was found to have a NSF on its antisense strand, and this gene is phylogenetically independent of the [Flavobacterium] nylB genes. Therefore, the presence of these NSFs is very rare and improbable. Even if the common ancestral gene of the nylB family was originally endowed with an NSF on its antisense strand, the probability of this original NSF persisting in one of its descendants of today is only 0.007. Unless an unknown force was maintaining the NSF, it would have quickly disappeared by random emergences of chain terminators. Therefore, the presence of such rare NSFs on all three antisense strands of the [three member] nylB gene family suggests that there is some special mechanism for protecting these NSFs from mutations that generate the stop codons. Such a mechanism may enable NSFs to evolve into new functional genes and hence seems to be a basic mechanism for the birth of new enzymes. [Emphasis added.]
Later on, they continue:
… the lifetime of a nonessential NSF is very short, and it is impossible for such a NSF to persist for a long period of evolution. Therefore, we strongly suggest that the existence of the NSFs on all the three antisense strands of the nylB gene family points to an unknown force that is preserving these nonessential NSFs; otherwise, they would have quickly disappeared by random emergences of chain terminators.
Ohno himself was aware of this work and in some sense supported it. He was the one who communicated it to the Proceedings of the National Academy of Sciences. What he made of it I don’t know.
The highlighted proposal in the above quotes is on the face of it antithetical to the materialist worldview. What kind of force can preserve apparently non-functional NSFs? Certainly a mechanism to preserve non-functional sequences so that they might some day evolve into functional genes is more suggestive of design than evolution. It would take a fair amount of foresight on the part of evolution, don’t you think, to develop a mechanism to prevent stop codons from interrupting non-functional NSFs, all for some possible future benefit?
All this speaks to the origin and preservation of potential information, information such as Ohno was looking for, but by a means different than he foresaw. We have returned full circle. Explaining nylonase does not require a frameshift, as I have shown in the first post — nonetheless nylonase’s gene is an unusual sequence. Getting overlapping code in three frames might happen in very rare circumstances, but keeping the NSFs open in the apparent absence of selection to maintain them would seem to be highly, highly unlikely. So we have extreme rarity piled upon rarity. Bear in mind also, that whatever the peculiar characteristics of the nylB gene sequence, it must also encode a functional, stably folded enzyme, which is another constraint.
Why am I going on and on about nylonase? It has to do with problem of the origin of novelty. Are frameshifts a possible source of new functional information? Might a sequence with alternate frames stay open by chance or be created by chance over evolutionary time? It’s a highly improbable event, but not impossible, I suppose. Might the alternate frames someday be material for frameshifted novel proteins, provided they stay open? They might theoretically be a reservoir for future proteins, but given what we know about the rarity of these kinds of sequences andthe rarity of protein folds in sequence space, the possibility of generating an entire new protein foldfrom a frameshift is extremely, extremely, extremelylow, and would depend on a highly unusual starting sequence tailored in advance for a particular functional specificity. In other words it would need to be designed.
In addition, even should such a sequence exist, it would not long persist in the face of neutral evolution. According to neo-Darwinism there is no magic molecular bouncer who throws out inactivating mutations before they can do their damage to a potential gene. Or to use another metaphor, evolution does not bank potentially useful sequences for future use. For it to do so would require foresight, an idea antithetical to evolutionary theory. Thus, any putative frame-shifted sequences that have been shown to have a functional role are better explained by design than by chance and necessity.
Should anyone disagree with my argument above, I’d like to point out that for a long time it was the standard belief among evolutionary biologists (and geneticists) that random sequence could not generate a functional protein. Frameshifted proteins are almost universally disrupted by stop codons (unless they happen to have an NSF or two like nylB). And even if they aren’t interrupted, the new sequence will be unlikely to fold into a stable protein, given the rarity of functional folds in sequence space (see the first post).
As an aside, as one of the curious facts of history, the disruptive properties of frameshift mutations were used to discover the triplet nature of the genetic code. Says Sir F.H.C. Crick in a lecture on the genetic code he gave in 1964:
This [the ability to combine mutations] has enabled us to tackle the question: is it really a group of three that makes up a codon? The basic idea is the following. We are able to pick up mutants which we believe (from the way they behave in various contexts) are not merely the change of one base into another, but are either the addition or a deletion of a base or bases. What happens when you have a genetic message and you put in an extra base? The reading starts from the beginning until it comes to that point and from there onward the whole of the message is read incorrectly, because it is being read out of phase. In fact we find that these [frameshift] mutants are completely inactive — this is one of our bits of evidence that they are what we say they are. You can pick up a number of such mutants and can put them together, by genetic methods, into the same gene. For example you can put together two of them. Such a gene would be read correctly until it reached the first addition, and then it would be out of phase. When the reading came to the second addition it would [be] read out of phase again, and so the whole of the rest of the message would be read incorrectly. Now it so happens that the left-hand end of this gene is not terribly important for its function. We can actually delete it and the gene will work after a fashion. In this region we have constructed, by genetic methods, a triple mutant, using three mutants all of the same type, and we have found that the gene will nevertheless function fairly normally.
This result is really very striking. Each of the three different faults, used singly, will knock out the gene. You can put them together in pairs in any combination you like, but then the gene is still quite inactive. Put all three in the same gene and the function comes back. We have been able to do this with a number of distinct combinations of three mutants (Crick et al., 1961).
Crick and others found that when three single base frameshift mutations of a particular gene, each completely disruptive on its own, were combined into the same gene, the three insertions together restored the frame enough for the protein to function again! Hence the code must be based on threes.
The sheer improbability of getting a functional enzyme from frameshifted random sequence has been the accepted view for a long time. It is only recently, in the era of big genomic data, that it has begun to be accepted that new proteins dooccasionally arise by frame-shift mutation. The reason? It’s because we find examples in the genome that appear to be products of such events, based on sequence comparisons.
The proteins apparently affected by such frameshifts in the genome are often transcription factors or membrane proteins involved in gene regulation. The apparent frameshift often affects alternative splicing and changes the coding sequence over an exon or so; alternatively, the frameshift affects the end of the protein, resulting in truncation. The fact that such a mutation is located near the protein’s end reduces the amount of disruption to the protein. Many such mutations have been documented to cause disease, however. For a demonstration, just use Google Scholar to search for “frameshift.”
At this point, the chief question that should be in everyone’s mind is, “Can evolution by neo-Darwinian means produce new functional information from frame-shifted sequence? Or are other explanations more likely?”
It boils down to this. Do we say that frameshifted functional proteins are easy to generate, because after all, they exist? Or do we acknowledge that such proteins are not easy to generate and so may be evidence for design?
To reiterate, it used to be standard knowledge that frameshift mutations were always bad. Disruptive. So, for example:
More radical mutational events, such as insertions and deletions that change the reading frame — frameshift mutations — are generally considered to be detrimental (e.g. by causing nonfunctional transcripts and/or proteins, through premature stop codons) and of little evolutionary importance, because they seriously alter the sequence and structure of the protein.
But now it has become popular to offer frameshifts as a quick way to get novelty. I am pretty sure it all began with Ohno, who said:
It has recently occurred to me that the gene started from oligomeric repeats at its certain stage of degeneracy (base sequence diversification) [nylB] can specify a truly unique protein from its alternative open reading frame.
Now the meme has spread. From the Abstract of a paper documenting the “Frequent appearance of novel protein-coding sequences by frameshift translation,” we hear that “Major novelties can potentially be introduced by frameshift mutations and this idea can explain the creation of novel proteins.” And how do they defend the possibility of a functional frameshift? “Some cases of recent evolution of new genes via frameshift have been reported. For example, in bacteria the sudden birth of an enzyme that degrades manmade nylon oligomers was explained by a frameshift translation of a preexisting coding sequence.”
Let us close by considering the nature of the argument being made concerning proposed frameshifts. The fact concerning such proposed frameshifts is that there are sequence similarities between two stretches of DNA, where one part appears to be frameshifted with respect to the other.
Notice that the argument used to explain the appearance of novel genes by frameshift uses a form of inference known as abduction, where one reasons from present effects to past causes.
The surprising fact A is observed.
If B were true, then A would be a matter of course.
Hence, there is reason to suggest that B is true.1
In other words:
The surprising fact of novel genes apparently arising by frameshift is observed.
If it is easy to get new functions from random sequence, then it is a matter of course that frameshifts can produce functional proteins.
Hence it is easy to get new functional proteins from random sequences
Abductive arguments are very weak. The problem is that there can be multiple competing causes that explain the observed effects. The only way to strengthen the argument is to rule out all other competing causes. And design is a particularly strong competing hypothesis. We know design is a cause capable of producing the effect in question, namely the generation of new functional proteins by the addition of frame-shifted code. In fact, given what we know about the rarity of functional proteins in sequence space, as demonstrated experimentally here, here, and here, and theoretically here, design is a betterexplanation than the neo-Darwinian one.
Until someone demonstrates experimentally, in real time, that a frameshift mutation can generate a new functional protein (not just a loss of function) by undirected processes, the inference that it is easy to do so is unjustified. And nylonase is not that demonstration.2
References:
(1) Stephen C. Meyer, Of Clues and Causes: A Methodological Interpretation of Origin of Life Studies. PhD dissertation (Cambridge: Cambridge University, 1990). Charles S. Peirce, “Abduction and Induction,” In The Philosophy of Peirce, edited by J. Buchler (London: Routledge, 1956), 150–154. Charles S. Peirce, Collected Papers, edited by Charles Hartshorne and P. Weiss. 6 vols. (Cambridge, MA: Harvard University Press, 1931–1935).
(2) In a future post, I will discuss experiments that attempt to demonstrate that random sequence can perform simple functions.
Editor’s note: Nylon is a modern synthetic product used in the manufacturing, most familiarly, of ladies’ stockings but also a range of other goods, from rope to parachutes to auto tires. Nylonase is a popular evolutionary icon, brandished by theistic evolutionist Dennis Venema among others. In a series of three posts, of which this is the second, Discovery Institute biologist Ann Gauger takes a closer look.
In an article yesterday, “The Nylonase Story: When Imagination and Facts Collide,” I described how some biologists claim that the enzyme nylonase demonstrates that it is easy to get new functional proteins. It has been proposed that nylonase is the result of a frameshift mutation that produced an entirely new coding sequence from an alternate reading frame. I showed why such a claim is false. Now I will explain what that means and something about the unusual properties of the nylB gene that caught molecular geneticist and evolutionary biologist Susumu Ohno’s attention.
What are alternate reading frames? To answer that question, I first need to provide some background information. I will begin by defining some terms I used in yesterday’s post. DNA is composed of two anti-parallel strands of nucleotides. The order of the nucleotides in each strand is what specifies the information the DNA carries. The two strands, called the sense and antisense strands, run in opposite directions. Even though their sequences are complementary, with A always paired with T, and C with G, each strand carries different potential information.
ATG GCA TGC ACC GGC ATT AG → sense TAC CGT ACG TGG CCG TAA TC ← antisense
Before the information in DNA can be used, it must be copied into what we call messengerRNA. The sequence of one strand of DNA, usually the sense strand, is copied using the same base complementarity: G pairs with C, and A with U (U is used in place of T in RNA). We call that copying transcription. The message that has been transcribed from the DNA into that sequence of RNA is now ready to be translated into protein.
Notice the language of information shot throughout these processes. The names for these processes were given by men fully committed to a naturalistic worldview, men such as Francis Crick and Sydney Brenner. Indeed, they were materialists one and all. Yet they saw the parallels between these processes and the human manipulation of text (language) or code (another form of language). The geneticcode is the framework that determines the relationship between groups of nucleotides (codons), and the amino acids they specify. The code specifies how to translate the messenger RNA that has been copied or transcribed from the DNA, so that it can be translatedinto a new language, the language of proteins. Below is an illustration of the standard genetic code (source here, used with permission):
Notice that the information in DNA is read in groups of three nucleotides (each group is called a codon), and each codon specifies a particular amino acid. Sometimes more than one codon can specify the same amino acid. For example in the top left corner, the table shows that UUU and UUC both specify the amino acid phenylalanine.
The nature of the code is such that it matters where the first codon begins — the first codon to be read establishes the codon groupings going forward. In the table above the “start” codon is AUG (it also specifies the amino acid methionine). The sequence of codons is “read” by a cellular machine called the ribosome, which starts reading the RNA message at AUG, and then proceeds three nucleotides at a time to translate the message into amino acids. In the sequence below, for example, the first codon to be read would be AUG and that codon determines the frame in which of all the other codons are read.
AUG GCA UGC ACC GGC AUU AGU
Now here’s where it gets interesting. Potentially, DNA can be grouped into different codons, or frames, depending on where the ribosome starts reading. See below for an illustration. For example, the sequence could potentially be read with the groupings shown in frame one (ATG GCA etc.) or frame two (TGG CAT etc., if a proper ATG exists somewhere upstream), leading ultimately to a different amino acid sequences for each. In fact there are six possible ways to group the DNA into codons — three frames on the sense strand going left to right (labeled 1-3), and three frames on the antisense strand (labeled 4-6), going right to left. Below I have laid out the six possible frames for the sequence we began with, but with the alternate frames staggered, and the alternate codons separated by spaces. Notice the sequence stays the same — the only thing that changes from frame to frame is how the nucleotides are grouped. It’s the same sequence, but it could be read and translated differently in each frame. This is because each codon specifies a particular amino acid. Thus, each frame results in a completely different string of amino acids.
frame 1 ATG GCA TGC ACC GGC ATT AG frame 2 TGG CAT GCA CCG GCA TTA G frame 3 GGC ATG CAC CGG CAT TAG
The codons TAA, TAG, and TGA are stop codons — they specify where the gene ends and protein translation stops. (For extra credit, can you find any ATG or stop codons in the above frames? They are there in both the forward and reverse direction. For more extra credit, can you use the code table to translate different frames, and demonstrate that each frame encodes a different protein?)
So when Venema and others say that nylonase arose by a frameshift mutation that produced a novel protein 392 amino acids long, they are claiming that a completely new coding sequence with frame-shifted codons could generate a functional protein. How likely is that? Not very, given the rarity of functional proteins in sequence space (see my first post). And, as I have already shown in my first post, such an unlikely hypothesis is unnecessary. The nylB gene appears to be the product of a simple gene duplication followed by two stepwise mutations to increase nylonase activity.
There is something special about the nylonase gene’s sequence though, something very odd. nylB has multiple large, overlapping (alternate) open frames that lack stop codons.
How hard is it to get a gene with multiple reading frames?
Let me explain. Roughly one in twenty codons are stop codons. A random DNA sequence will have stop codons about every sixty bases, and may or may not have a start codon. Usually the alternate frames of DNA sequences are interrupted by stop codons. Only the frame that actually specifies the correct gene will have no stop codons at all over a significant length. This system is actually very ingenious. The one frame that needs to be read and translated is identified by an ATG. The other frames will usually lack an ATG and/or will have several stop codons that interrupt their translation, thus preventing the cell from wasting energy on nonsense transcripts.
According to the nylonase story, as told by Ohno and Venema and numerous others, a new ATG start codon was formed by the insertion of a T between an A and G, thus creating a new start codon after the original ATG, which shifted the reading frame for that sequence to that specified by the new ATG, and creating a completely different coding sequence and thus a new protein. Let us grant that scenario for the sake of argument. Normally such a shift would produce a new coding sequence that would be interrupted by stop codons, so the newly frameshifted protein would be truncated. Thus the only reason this frameshift hypothesis for nylonase is even remotely possible is because the sequence coding for nylonase is most unusual, and contains not one, not two, but three open frames Although frameshift mutations are ordinarily considered to be quite disruptive, at least in this case the putative brand new protein sequence would not terminate early due to stop codons.
My point? The first step to getting a new functional protein of any length from a frameshift is to avoid stop codons. The odds of a random coding sequence having an open alternate frame, without stops, are poor. As a consequence, if a protein does have an open frame in addition to its coding sequence, it’s worth paying attention to. And it so happens that nylonase does have more than one open frame. The DNA sequence above illustrates the six frames, numbering them frames 1 through 6. Using that convention, frames 1 and 3 are read from the sense strand. Both have no stop codons over the length of the gene in the sense direction. Frame 4 on the antisense direction has no stop codons either. Frame 1 is the coding frame that specifies the nylonase protein, otherwise known as the open reading frame (ORF). It is defined by the presence of both a start and stop codon. The other two frames have no start codons or stop codons, so I’ll call them non-stop frames (NSFs). They are frames 3 and 4.
The probability of a DNA sequence with an ORF on the sense strand and 2 NSFs is very small. Just exactly howsmall are the chances of avoiding a stop codon in three out of six frames? We set out to determine that by performing a numerical simulation using pseudorandom numbers to generate sequences at various levels of GC content. (By we I mean that my husband, Patrick Achey, who is an actuary, did the programming work, while I determined the parameters.) We chose to vary the GC content because sequences with a higher GC content have fewer stop codons. Remember, a stop codon always has an A and a T (TAA, TAG, and TGA are the stop codons) so having a sequence with a lower percentage of AT content will reduce the frequency of stop codons. Conversely, higher GC content makes the chances of avoiding stop codons and getting longer ORFs much greater, thus also increasing the chances of NSFs. The genomes of bacteria vary in their GC content, from less than 20 percent to as much as 75 percent, though the reason why is not known. One species of Flavobacteriumhas a genome with about 32 percent GC and 2400 genes — the precise values varies with the strain. The plasmid on which nylB resides is very different. It has 65 percent GC content. The gene encoding nylonase has an even higher 70 percent GC content, which is near the observed bacterial maximum of 75 percent.
We chose to use a target ORF size of 900 nucleotides (or 300 amino acids) because it is an average size for a functional protein. Nylonase is 392 amino acids long; the small domain of beta lactamase, the enzyme my colleague Doug Axe studied, is about 150 amino acids long. The median length for an E. coli protein is 278 amino acids; for humans, the median length is 375.
As expected, the simulation showed that the higher the GC content, the greater the likelihood that ORFs that are 900+ nucleotides long exist. At 50 percent GC, the average ORF length we obtained was about 60 nucleotides; most ORFs terminate well before 900 nucleotides. Indeed, in our simulation only two out of a million random sequences made it to 900 nucleotides before encountering a stop codon. As a result, we could not determine the rarity of NSFs at 50 percent GC — we would probably have to run the simulation for more than a billion trials to get any significant number of NSFs at all.
Sequences at 60 percent GC gave 57 ORFs at least 900 nucleotides long out of a million trials, while sequences at 65 percent GC produced 404 out of a million, one of which also had an NSF.
NSFs were much more probable for sequences that were 70 percent GC, like nylB. In our simulation 3,021 out of a million trials were ORFs at least 900 nucleotides long. That’s a frequency of .3 percent. Of those 3,021 ORFs, 86 had 1 NSF, and none had 2 NSFs. We had to run 10 million trials at 70 percent GC to see any ORFs with 2 NSFs. From those 10 million randomly generated sequences, we obtained 28,603 ORFs; 903 had 1 NSF and only 9 had 2 NSFs.
Interestingly, at 80 percent GC we got a few sequences with 4 NSFs; but I don’t know of any bacterium with a GC content that high.
Our simulation shows that multiple NSFs are very rare. The probability that an ORF 900 nucleotides long with 70 percent GC content will have two NSFs is 9 out of 28,603, or 0.0003. If these figures are recast to include the total number of trials required to get an ORF of that length and GC content and with 2 NSFs, the probability would be 9 out of 10,000,000 trials.
A sequence like nylB is very rare. In fact, I suspect that for all cases where overlapping genes exist, in other words where alternate frames from the same sequence have the potential to code for different proteins, unusual sequence will necessarily be found. Likely it will be high in GC content. Could such rare sequences be accidental? I think that if we compare the expected number of alternate or overlapping NSFs per ORF, with the actual number we will find that there are more of these alternate open reading frames than would be predicted by chance.
Thus, bacterial genomes contain a larger number of long shadow ORFs [ORFs on alternate frames] than expected based on statistical analysis. Random mutational drift would have eliminated the signal long ago, if no selection pressures were stabilizing shadow ORFs. Deviations between the statistical model and bacterial genomes directly call for a functional explanation, since selection is the only force known to stabilize the depletion of stop codons. Most shadow genes have escaped discovery, as they are dismissed as false positives in most genome annotation programs. This is in sharp contrast to many embedded overlapping genes that have been discovered in bacteriophages. Since phages reside in a long term evolutionary equilibrium with the bacterial host genome, we suggest that overlooked shadow genes also exist in bacterial genomes.
Indeed, a study of the pOAD2 plasmid from which nylB came indicates that there are potentially many overlapping genes on that plasmid. nylB′, for example, a homologous gene on the same plasmid that differs by 47 amino acids from nylB, also has 2 NSFs. These unusual and unexpected features of DNA have consequences for how we think about the origin of information in DNA sequences, as I shall discuss in the next post.
Monday: “The Nylonase Story: The Information Enigma.”
Editor’s note: Nylon is a modern synthetic product used in the manufacturing, most familiarly, of ladies’ stockings but also a range of other goods, from rope to parachutes to auto tires. Nylonase is a popular evolutionary icon, brandished by theistic evolutionist Dennis Venema among others. In a series of three posts, Discovery Institute biologist Ann Gauger takes a closer look.
A significant problem for the neo-Darwinian story is the origin of new biological information. Clearly, information has increased over the course of life’s history — new life forms appeared, requiring new genes, proteins, and other functional information. The question is — how did it happen? This is the central question concerning the origin of living things.
Stephen Meyer and Douglas Axe have made this strong claim:
[T]he neo-Darwinian mechanism — with its reliance on a random mutational search to generate novel gene sequences — is not an adequate mechanism to produce the information necessary for even a single new protein fold, let alone a novel animal form, in available evolutionary deep time.
Their claim is based on the experimental finding by Doug Axe that functional protein folds are exceedingly rare, on the order on 1 in 10 to the 77th power, meaning that all the creatures of the Earth searching for the age of the Earth by random mutation could not find even one medium-size protein fold.
In contrast, Dennis Venema, professor of biology at Trinity Western University, claims in his book Adam and the Genome and in posts at the BioLogos website that getting new information is not hard. In his book, he presents several examples he thinks demonstrate the appearance of new information — the apparent evolution of new protein binding sites, for example. But the best way to reveal Axe and Meyer’s folly, he thinks, (and says so in his book and a post at BioLogos) would be to show that a genuinely “new” protein can evolve.
…[E]ven more convincing… would be an actual example of a functional protein coming into existence from scratch — catching a novel protein forming “in the act” as it were. We know of such an example — the formation of an enzyme that breaks down a man-made chemical.
In the 1970s, scientists made a surprising discovery: a bacterium that can digest nylon, a synthetic chemical not found in nature. These bacteria were living in the wastewater ponds of chemical factories, and they were able to use nylon as their only source of food. Nylon, however, was only about 40 years old at the time — how had these bacteria adapted to this novel chemical in their environment so quickly? Intrigued, the scientists investigated. What they discovered was that the bacteria had an enzyme (which they called “nylonase”) that effectively digested the chemical. This enzyme, interestingly, arose from scratch as an insertion mutation into the coding sequence of another gene. This insertion simultaneously formed a “stop” codon early in the original gene (a codon that tells the ribosome to stop adding amino acids to a protein) and formed a brand new “start” codon in a different reading frame. The new reading frame ran for 392 amino acids before the first “stop” codon, producing a large, novel protein. As in our example above, this new protein was based on different codons due to the frameshift. It was truly “de novo” — a new sequence.
Venema is right. If the nylonase enzyme did evolve from a frameshifted protein, it would genuinely be a demonstration that new proteins are easy to evolve. It would be proof positive that intelligent design advocates are wrong, that it’s not hard to get a new protein from random sequence. But the story bears reexamining. Is the new protein really the product of a frameshift, or did it pre-exist the introduction of nylon into the environment? What exactly do we know about this enzyme? Does the evidence substantiate the claims of Venema and others, or does it lead to other conclusions?
First, some history. In the 1970s Japanese scientists discovered that certain bacteria had developed the ability to degrade the synthetic polymer nylon. Okada et al. identified three enzymes responsible for nylon degradation, and named them EI, EII, and EIII. The genes that encoded them were named nylA,nylB, and nylC. They sequenced the plasmid on which the genes were found, and discovered that there was another gene on the same plasmid that was very similar to nylB; they named it nylB′. (We will focus on the story of nylB and nylB′ because they are the ones relevant to Venema’s story.)
So far all I have given you are the facts. Now here’s the interpretation of these facts. Some claimed that the nylonase enzyme, as it was called, had originated some time after people began making nylon (in the 1930s). That seemed plausible because nylonase was unable to degrade naturally occurring amide bonds — it could degrade only the amide bonds in nylon — and so had not existed previously, it was thought. The popular conclusion was that the nylonase activity evolved in response to the presence of nylon in the environment, and thus was only forty years old. And here’s the big interpretive leap: it must not be hard to get new enzymes if a new one can evolve within a period of forty years.
Okada et al. had sequenced the genes encoding nylB and nylB′. They concluded that the nylonase activity was the result of a gene duplication followed by several mutations to the nylB gene. But at this point Susumu Ohno, an eminent molecular geneticist and evolutionary biologist, noticed something unusual about the nylB gene sequence (Ohno, 1984). Ohno had a theory that DNA with repeats of the right kind had the potential to code for protein in multiple frames, with no interrupting stop codons, and might thus be a source for “new” proteins. (If you are unfamiliar with the terms I just used, I invite you to take a look at my post tomorrow, where I will explain the necessary concepts. For those already familiar, I present some relevant data concerning the rarity of sequences that can be frameshifted.)
Ohno noticed that nylB, the gene for nylonase, might originally have encoded something else if a certain T was removed. The nylonase gene as it exists now has 1179 bases, which encode a 392 amino acid protein. Without a particular T embedded in the ATG start codon, though, the sequence would have specified a hypothetical original gene with a longer open reading frame (ORF) of 427 amino acids, in a different frame. Thus, Ohno proposed a “new” protein with a new function acting on a new substrate was born when a T inserted in between a particular A and G in the DNA, making a new ATG start codon and shifting the frame to code for a new protein, the protein we now call nylonase.
Ingenious. According to Ohno, nylonase could be a new enzyme, appearing suddenly with no known precursors via a sudden frameshift. (Note that all of this assumes that new protein folds are easy to get.) Ohno published this hypothesis in the Proceedings of the National Academy of Sciences. It was a hypothesis only, however, as a careful reading of his paper shows. One heading, for example:
R-IIA Coding Sequence [nylB] for 6-AHA LOH [nylonase] Embodies an Alternative, Longer Open Reading Frame That Might Have Been the Original Coding Sequence [Emphasis added.]
and the text says:
I suggest that the RS-IIA base sequence [nylB] was originally a coding sequence for an arginine-rich polypeptide chain 427 or so residues long in its length and that the coding sequence for one of the two isozymic forms of 6-ALA LOH [nylonase] arose from its alternative open reading frame. [Emphasis added.]
Ohno presented arguments for why his suggestion was plausible, but did not provide evidence that the “original” gene ever existed or was used (in fact he says it was unlikely to be useful based on its amino acid composition), or that the insertion ever happened. Nonetheless, the frame-shift hypothesis for the origin of nylonase has been widely proclaimed as fact (though, notably, not by Okada et al. who have done most of the work).
If the nylonase story as told above were true, namely that a frameshift mutation resulted in the de novo generation of a new protein fold with a new function, it would indeed constitute a substantial refutation to Meyer and Axe’s claim. If a frame-shift mutation can produce a random new open reading frame in real, observable time, and give rise to a new functional enzyme, then it must not be that hard to make new functional protein folds. In other words, functional protein folds must not be rare in sequence space. And therefore Stephen Meyer’s arguments about the difficulty of getting enough new biological information to generate a new fold must be wrong as well. Venema flatly asserts:
If de novo protein-coding genes such as nylonase can come into being from scratch, as it were, then it is demonstrably the case that new protein folds can be formed by evolutionary mechanisms without difficulty….[I]f Meyer had understood de novo gene formation — as we have seen, he mistakenly thought it was an unexplained process — he would have known that new protein folds could indeed be easily developed by evolutionary processes.
Slam dunk, right?
A little caution in accepting this story without hard evidence would be wise. In genetics we are taught that frame-shift mutations are extremely disruptive, completely changing the coding sequence and resulting in truncated nonsense. In fact, one term for a frameshift mutation is “nonsense mutation.” A biologist’s basic intuition should be that frameshifts are highly unlikely to produce something useful. The only reasons for the widespread acceptance of Ohno’s hypothesis that I can come up with are the unusual character of the sequence itself, Ohno reputation as a brilliant scientist (which he was), and wish-fulfillment on the part of some evolutionary biologists.
Fortunately, science marches on, and evidence continues to accumulate. The same group of Japanese scientists continued their study of the nylonase genes. nylB appeared to be the result of a gene duplication of nylB′ that occurred some time ago. EII′ (the enzyme encoded by nylB′) had very little nylonase activity, while EII (the enzyme encoded by nylB) was about 1000 fold higher in activity. The two enzymes differed in amino acid sequence at 47 positions out of 392. With some painstaking work, the Japanese determined that just two mutations were sufficient to convert EII′ to the EII level of activity.
They then obtained the three-dimensional structure of an EII-EII′ hybrid protein. And with those results everything changed — or should have.
…the three-dimensional structure of the protein has been solved using X-ray crystallography, a method that gives us the precise shape of the protein at high resolution. Nylonase is chock full of protein folds— exactly the sort of folds Meyer claims must be the result of design because evolution could not have produced them even with all the time since the origin of life. [Emphasis added.]
Unfortunately, Venema doesn’t have the story straight. Nylonase has a particular fold, a particular three-dimensional, stable shape. Most proteins have a distinct fold — there are several thousand kinds of folds known so far, each with a distinct topology and structure. Folds are typically made up of small secondary structures called alpha helices and beta strands, which help to assemble the tertiary structure — the fold as a whole. Venema seems unclear about what a protein fold is, and the distinction between secondary and tertiary structures. Nylonase is not “chock full of folds.” No structural biologist would describe nylonase as “chock full of protein folds.” Indeed, no protein is “chock full of folds.” Perhaps Venema was referring to the smaller units of secondary structure I mentioned above, the alpha helices or beta strands. But it would appear he doesn’t know what a protein fold is.
Maybe that explains why Venema missed the essential point of the paper describing nylonase’s structure. The crystal structure of EII-EII’ (a nylonase hybrid necessary to be able to crystalize the protein) revealed that it is not a new kind of fold, but a member of the beta-lactamase fold family. More specifically, it resembles carboxylesterases, a subgrouping of that family. In addition, when the scientists checked EII′ and EII, they found that both enzymes had previously undetected carboxylesterase activity. In other words, the EII’ and EII enzymes were carboxylesterases. If it looks like a duck and quacks like a duck, it is a duck.
Thus, EII′ and EII did not have frameshifted new folds. They had pre-existing folds with activity characteristic of their fold type. There was no brand-new protein. No novel protein fold had emerged. And no frameshift mutation was required to produce nylonase.
Where did the nylon-eating ability come from? Carboxylesterases are enzymes with broad substrate specificities; they can carry out a variety of reactions. Their binding pocket is large and can accommodate a lot of different substrates. They are “promiscuous” enzymes, in other words. Furthermore, the carboxylesterase reaction hydrolyzes a chemical bond similar to the one hydrolyzed by nylonase. Tests revealed that both the EII and EII′ enzymes have carboxylesterase and nylonase activity. They can hydrolyze both substrates. In fact it is possible both had carboxylesterase activity anda low level of nylonase activity from the beginning, even before the appearance of nylon.
nylB′ may be the original gene from which nylB came. Apparently there was a gene duplication at some point in the past. The two genes appear to have acquired mutations since then — they differ by 47 amino acids out of 392. The time of that duplication is unknown, but not recent, because it takes time to accumulate that many mutations. However, at least some of those mutations must confer a high level of nylonase activity on EII, the enzyme made by nylB. The enzyme EII’ made by nylB’ has only a low ability to degrade nylon, while EII degrades nylon 1000 fold better. So one or more of those 47 amino acid differences must be the cause of the high level of nylonase activity in EII. Through careful work, the Japanese workers Kato et al. identified which amino acid changes were responsible for the increased nylonase activity. Just two step-wise mutations present in EII, when introduced into EII’, could convert the weak enzyme EII’ to full nylonase activity.
Our studies demonstrated that among the 47 amino acids altered between the EII and EII’ proteins, a single amino acid substitution at position 181 was essential for the activity of 6-aminohexanoate-dimer hydrolase [nylonase] and substitution at position 266 enhanced the effect.
So. This is not the story of a highly improbable frame-shift producing a new functional enzyme. This is the story of a pre-existing enzyme with a low level of promiscuous nylonase activity, which improved its activity toward nylon by first one, then another selectable mutation. In other words this is a completely plausible case of gene duplication, mutation, and selection operating on a pre-existing enzyme to improve a pre-existing low-level activity, exactly the kind of event that Meyer and Axe specifically acknowledge as a possibility, given the time and probabilistic resources available. Indeed, the origin of nylonase actually provides a nice example of the optimization of a pre-existing fold’s function, not the innovation or creation of a novel fold.
Here, we propose that amino acid replacements in the catalytic cleft of a preexisting esterase with the beta-lactamase fold resulted in the evolution of the nylon oligomer hydrolase. [Emphasis added.]
Let’s put to bed the fable that the nylon oligomer hydrolase EII, colloquially known as nylonase, arose by a frame-shift mutation, leading to the creation of a new functional protein fold. There is absolutely no need to postulate such a highly improbable event, and no justification for making this extravagant claim. Instead, there is a much more parsimonious explanation — that nylonase arose by a gene duplication event some time in the past, followed by a series of two mutations occurring after the introduction of nylon into the environment, which increased the nylon oligomer hydrolase activity of the nylB gene product to current levels. Could this series of events happen in forty years? Most certainly. Probably in much less time. In fact, it has been reported to happen in the lab under the right selective conditions. And most definitely, the evolution of nylonase does not call for the creation of a novel protein fold, nor did one arise. EII’s fold is part of the carboxylesterase fold family. Carboxylesterases serve many functions and have been around much longer than forty years.
Douglas Axe and Stephen Meyer readily admit that this kind of evolutionary adaptation happens easily. A protein that already has a low level of activity for a particular substrate can be mutated to favor that side reaction over its original one, often in just a few steps. There are many cases of this in the literature. What Axe and Meyer do claim is that generating an entirely new protein fold via mutation and selection is implausible in the extreme. Nothing in the nylonase story that Dennis Venema tells shows otherwise.
Tomorrow: “The Nylonase Story: How Unusual Is That?”
Photo: Nylon parachute, by Lance Corporal Brian D. Jones, U.S. Marine Corps [Public domain], via Wikimedia Commons.
There is a whole school of research aimed at showing we are not so separate from chimps, and even that they are already on the way up. In a recent BBC article by Colin Barras the focus was on the use of stone tools, and the bold claim that chimps and some monkeys are already in the Stone Age because of their use of rocks as tools.
Some of the reports of stone use appear valid. Capuchins and chimpanzees are all known to use stones to crack open food, and the technique appears to go back thousands of years. But then, no one is disputing that some animals use simple tools. Even otters use stones to break open clam shells.
Then there is the social side. Another BBC article, this one by Melissa Hogenboom, describes ways that chimps show empathy and social awareness, the ability to read facial expressions, to hide things from others, and to share. But then those things are not so unique–anyone who has lived with dogs knows how acutely sensitive to social cues and facial expressions they are, and how they seem to show guilt over bad behavior. They can even share–or be greedy, depending on the dog’s temperament.
And of course, she describes how chimps have the capacity for language. Translation: chimps that have lived and trained with humans can have a vocabulary of 500 words, and can understand thousands more.
But is this a sign they are on the road to becoming like us?
In a further BBC article, Hogenboom lays out the argument against the idea that there is little difference between us and chimps. She quotes Ian Tatersall, a paleoanthropologist at the American Museum of Natural History in New York: “Obviously we have similarities. We have similarities with everything else in nature; it would be astonishing if we didn’t. But we’ve got to look at the differences,”
Hogenboom writes:
When you pull together our unparalleled language skills, our ability to infer others’ mental states and our instinct for cooperation, you have something unprecedented. Us.
‘Just look around you,’ Tomasello [of the Max Planck institute for Evolutionary Anthropology in Leipzig, Germany,] says, ‘we’re chatting and doing an interview, they (chimps) are not.’
We have our advanced language skills to thank for that. We may see evidence of basic linguistic abilities in chimpanzees, but we are the only ones writing things down.
We tell stories, we dream, we imagine things about ourselves and others and we spend a great deal of time thinking about the future and analyzing the past.
Not to mention the Brandenburg Concertos. Calculus. Hamlet. The Pythagorean theorem. The discovery of the periodic table. Einstein’s theory of relativity. All works of genius.
In her first article I think Hogenboom overplays the similarities between chimps and humans, just as Barras exaggerates their tool-making ability. Both authors accept the standard Darwinian view, however. Perhaps that accounts for their need to have a story of underlying similarity.
Ms. Hogenboom attributes all this to our big brains in her second article. She does admit that nobody knows how our brains came to be big. Neither does anyone have any solid explanation for how the anatomical, developmental, and physiological differences between us and chimps came to be.
At least Ms. Hogenboom acknowledges there might be a difficulty in explaining things.






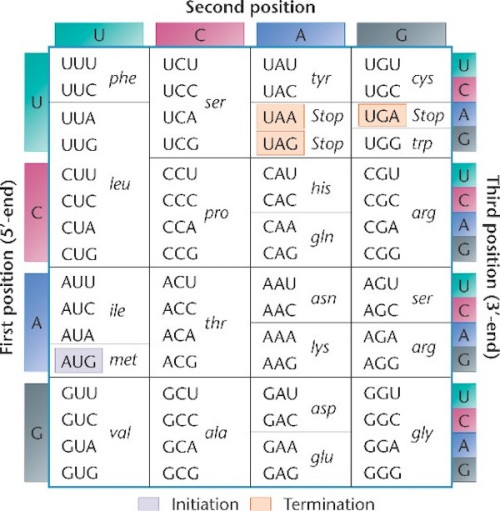

{kind=link}
{kind=link}
{kind=link}